Step 7 Methylation databases
To analyze the methylation pattern of the ACE2 gene, we selected BECon: A tool for interpreting DNA methylation findings from blood in the context of the brain, and DiseaseMeth version 2.0 - The human disease methylation database.
The aim of BECon (Blood-Brain Epigenetic Concordance) is to allow for improved interpretation of surrogate methylation results by looking at the relationship between human blood and brain methylation. Spearman correlation values of methylation between blood and the listed brain region are shown below. In these columns, colors are used to show the percentile of all correlations each value reaches (see legend). Darker grey and orange represent lower or higher correlations, respectively. The cell composition metrics are the difference between betas for unadjusted and adjusted data. So how much the higher the beta values, the higher the amount of methylated CpG.
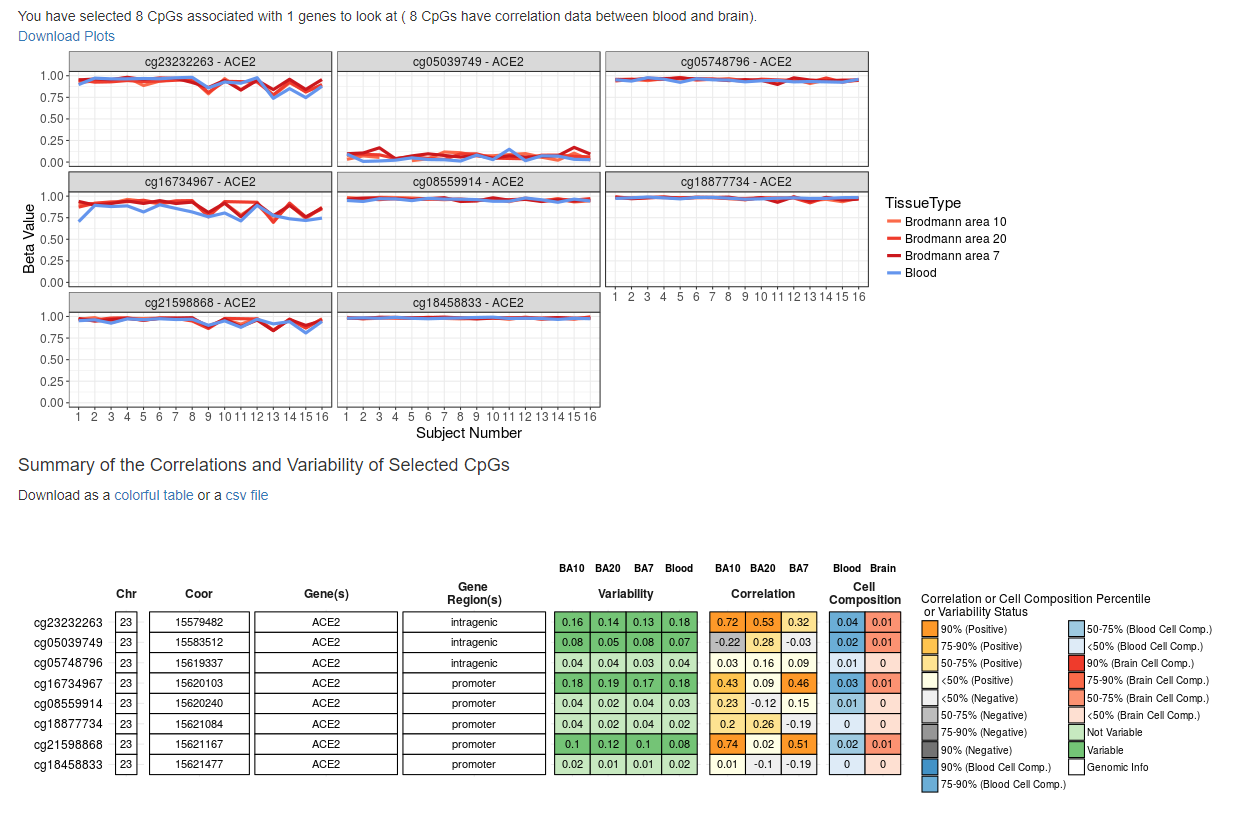
We can observe that the ACE2 gene shows a similar pattern of CpG context across samples and tissues (in the context of blood and brain).
To access the methylation pattern of ACE2 in a cancer context, we used DiseaseMeth. The Search Engine has three query possibilities: GeneSearch (queries DNA methylation level for disease-associated genes by disease type), DiseaseSearch (queries DNA methylation level across diseases for a specified gene/genomic position), AdvanceSearch (queries disease methylome or DNA methylation level for a specified gene/genomic position).
- Disease option is used to select interesting diseases. We selected all the cancer types and diseases in order to compare the results.
- Gene name/Transcript ID/Genomic Interval is used to set interesting genomic regions. Users can input multiple items. We selected ACE2 only.
- The platform option is used to select the data type of methylome. We selected all the methylation platforms.
- Controls from the same tissue/cell line are used to select all controls from different datasets but the same tissue/cell line instead of the option of controls from the same dataset (as in some experiments there are no controls matched with cases).
- Differential Analysis method: select the method for case-control differential analysis. Comparison can be performed by t-test or edgeR. We used the default t-test.
- It is necessary to set a p-value for the analysis. The results will be marked with red color in the results panel. The options are 0.01 and 0.05 and we selected 0.05.
- Absolute methylation difference: this option is used to filter the differentially methylated region together with the p-value.
The result will display the outcome of differential analysis, disease-gene association, and methylation profile of the given region in the selected diseases. The differential analysis will be applied in the TSS or genomic region by t-test between cases and controls. Survival analysis and functional annotation can also be applied. The Gene-Gene Relationship Analysis and Disease-Disease Relationship Analysis will display the Pearson correlation coefficient between genes or diseases.
Differential methylated results - case vs. control: Here, we provide 3 examples of the query results:
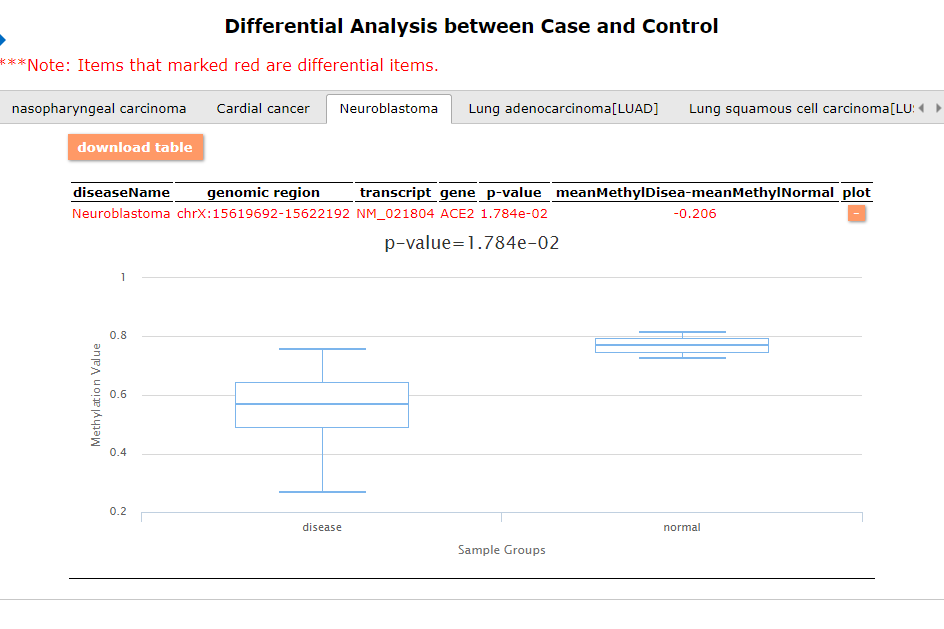
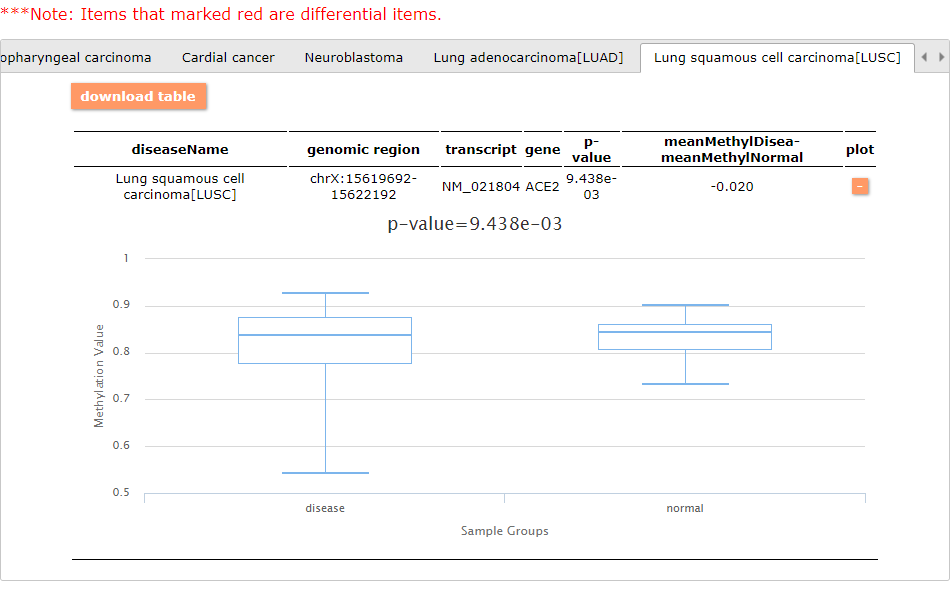
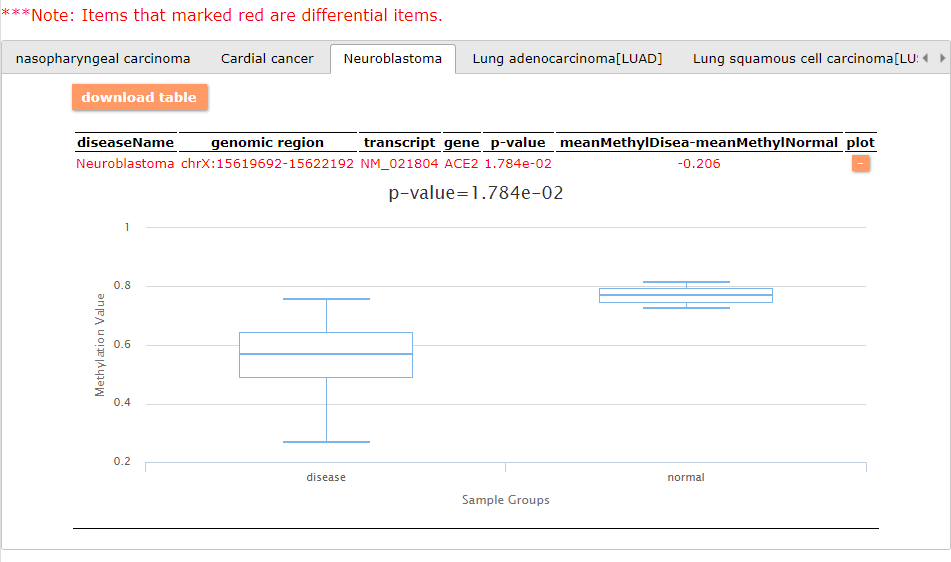
In general, the ACE2 expression in the cancer samples are smaller than in the normal samples. This may indicate the role of the enzyme in the tumoral context.